Air Care Series: Long QT Syndrome
/History of Present Illness
A pediatric male with a history of congenital long QT syndrome (LQTS) involving mutation of the LQT2 gene on daily propranolol presented after reportedly having an episode of unresponsiveness at home. The patient’s mother stated that the patient was previously in his usual state of health and received his dose of propranolol the prior night. Upon awakening that morning, the patient was sweaty, cold to the touch, and irritable. He then started to stare off into space, subsequently becoming unresponsive with sudden loss of tone and tonic-clonic movements.
Upon EMS arrival, the patient had a blood glucose in the 30s and was treated with intramuscular glucagon and parenteral dextrose. Subsequent repeated finger stick blood glucose remained in the 30s and Air Care was called to the scene.
Past Medical History
Long QT Syndrome
Past Surgical History
Bilateral Myringotomy
Medications
Propanolol
Allergies
None
Vitals
Temp HR BP RR SpO2
35C 52 80/54 30 99%RA
Physical Exam
Age appropriate two-year-old male who is obtunded and not responsive. His lungs were clear bilaterally and his heart had a regular rate and rhythm. His abdomen was soft and non-distended. His extremities were cool to the touch with delayed capillary refill. Bruising and edema was present on RLE at the site of the attempted IO placement. His pupils were equal and reactive, he did not respond to voice or follow commands, and did not localize to central painful stimuli or withdraw from peripheral painful stimuli.
Diagnostic Work-Up
VBG: pH7.20 pCO2 60 HCO3 26 BE-4
Ca 9 Phos 5 Alb 4
Urine: +ketones, +glucose, otherwise normal
EKG: Normal sinus rhythm at 63 bpm, normal axis, normal intervals: PR 140 ms, QRS 62 ms, QTC 433 ms.

Hospital Course
Upon Air Care arrival, the patient was bradycardic in the 50s with otherwise normal vital signs, and unresponsive. The medical crew administered a second dose of parenteral glucose and the patient became more responsive but still drowsy and confused. A repeat finger stick glucose was 349. He received a dose of ondansetron and was warmed with blankets. He remained stable throughout the flight and was transported to a quaternary children’s hospital in stable condition.
In the emergency department (ED), the patient was initially hypothermic and warmed with a Bair Hugger, blankets, and warmed IV fluid bolus. He intermittently had bradycardia to the 50s, which improved with stimulation. The patient had blood cultures drawn and was started on ceftriaxone for empiric sepsis coverage. The patient’s respiratory acidosis was thought to be due to hypoventilation from a postictal period after a presumed seizure. Beta-blocker overdose remained high on the differential, although the patient’s parents were adamant that the patient did not receive any more than his usual dose. The patient was admitted to the pediatric intensive care unit (PICU) for continuous monitoring and further workup.
The patient was evaluated by cardiology and endocrinology during his hospitalization. His EKG in the emergency department revealed a rate of 63 and a normal QTc of 435ms, both of which were likely due to continuation of his propranolol. He had no QTc prolongation, arrhythmias, or seizure-like episodes. His heart rate remained in the 70s-110s during his hospitalization and he was continued on his home dose of propranolol.

Figure 1: Graph depicting the cardiac action potential in association with QT interval
The etiology of his hypoglycemia was not definitively identified during his initial hospitalization but thought to be secondary to ketotic hypoglycemia versus decreased oral intake in setting of viral illness worsened by blunting of his body’s natural response to hypoglycemia by propranolol. There are also reports of congenital LQTS mutations being associated with abnormal glycemic regulation. He was subsequently admitted for a hypoglycemia challenge and diagnosed with ketotic hypoglycemia. His seizure was thought to be secondary to his hypoglycemia.

Table 1: Upper limit of QT interval values
Discussion:
While this patient’s hospitalization primarily focused on diagnsosis and treatment of his hypoglycemia, the discussion to follow will focus on the pathophysiology and management of LQTS and the malignant arrhythmias that may develop in these patients.
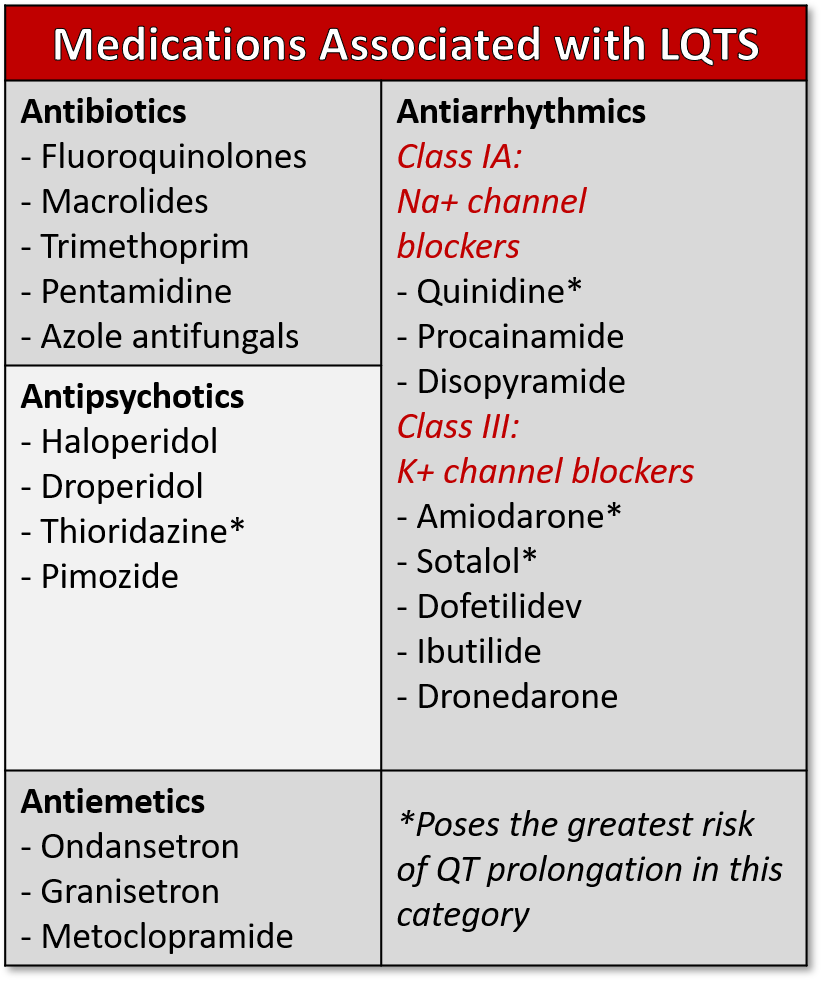
Table 2: Medications associated with prolonged QT syndrome
LQTS is an EKG abnormality which can be congenital or acquired, and is characterized by lengthening of the QT interval as measured from the start of the QRS complex to the termination of the T-wave, which represents the depolarization-repolarization cycle of the ventricles. LQTS is generally a disorder of repolarization rather than depolarization. Depolarization occurs through the influx of sodium and calcium, and repolarization occurs through the outflow of potassium through channels in the myocardial membrane. (1) (Figure 1) When corrected for heart rate (called the QTc interval), the upper limit of normal for prepubertal children is 460ms, 470ms in adult males, and 480ms in adult females. (2) Lengthening of the QTc beyond these durations can lead to Torsades de Pointes or ventricular fibrillation, which can subsequently lead to syncope or even sudden cardiac death. A meta-analysis performed by Zhang et al found that QT-interval length is a determinant of mortality in the general population. (3)

Table 3: Risk factors for developing prolonged QT
Congenital LQTS is estimated to have a prevalence of approximately 1:2000. (5) Mutations in ten genes have been linked to congenital long QT. However, the best described mutations are LQT1 and LQT2, which are mutations in potassium channels, and LQT3 which is a mutation in a sodium channel. Together these mutations comprise over 75% of the cases of congenital LQTS. The penetrance of these mutations is highly variable and some patients will have normal QTc intervals and remain completely asymptomatic throughout life even when they test positive for the mutations. Sudden cardiac death, however, was the initial manifestation in 13% of cases in a retrospective review. (13)
Causes of acquired LQTS include hypokalemia, hypocalcemia, hypomagnesemia, and drugs. Many drugs (table 3) have been implicated in lengthening of the QT interval, but class IA and III antiarrhythmics and psychotropic medications are the most common medications that prolong the QT interval. (6) Electrolyte abnormalities or drugs that prolong the QT interval can unmask LQTS in patients with a genetic predisposition who otherwise may have been asymptomatic.
Typically, patients who present to the emergency department that are symptomatic from LQTS will have a history of palpitations, syncope, presyncope, seizures, or cardiac arrest. (4) Classic triggers for symptoms include exercise, emotional stress, diving or swimming and loud loud noises. (4) Patients who are seen in the emergency department reporting syncope or seizure activity should have an EKG to screen for LQTS. While the EKG machine calculates the QTc, the QT interval needs to be closely inspected and manually calculated if it appears prolonged. Leads II and V5 are conventionally used to measure the QTc. If a U wave is present, this should not be included when measuring the QT interval. A QTc greater than 480ms in pediatric patients or 500ms in adults on a screening EKG needs to be further evaluated. Any culprit drugs should be discontinued and metabolic abnormalities must be corrected aggressively. Risk factors for LQTS should be carefully assessed prior to administering a QT-prolonging medications in the emergency department (Table 3). (1) If a patient presents with unstable Torsades de Pointes, the treatment is defibrillation; if stable, they should receive intravenous magnesium sulfate and overdrive transvenous pacing if persistent. (6,7) Several studies have shown that prophylactic administration of magnesium to patients requiring a QT-prolonging medication can decrease dysrhythmias and shorten the QTc, and this may be considered in patients with LQTS risk factors who absolutely require continued administration of QT-prolonging medications. (1)
A careful family history should be taken and patients should be referred to cardiology if there is concern or congenital LQTS. Beta-blocker therapy, specifically propranolol and nadolol, has a proven mortality benefit in patients with congenital LQTS. If determined to be at high risk (e.g. survivors of cardiac arrest, syncope despite maximum beta blocker therapy, QTc >550 ms), patients may require left cardiac sympathetic denervation and/or implantable cardiac defibrillator. (8)
LQTS may be congenital or acquired and is most often found on screening EKG, but occasionally presents as sudden arrhythmias or cardiac arrest. Providers must maintain a high index of suspicion and consider the diagnosis in all cases of syncope or cardiac arrest, particularly in the young patient. Treatment ranges from beta-blockade for the asymptomatic to ICD placement for those symptomatic. Avoidance of QT prolonging medications is also essential to prevent degeneration to Torsades or life threatening arrhythmias. Patients should be screened for electrolyte abnormalities and these should be aggressively corrected. LQTS is a life threatening condition and patients who presents with symptoms and a QTc greater than 500ms should be admitted for observation and further management, while those who are asymptomatic should have close follow up arranged.
References
Pourmand A, Mazer-Amirshahi M, Chistov S, Sabha Y, Vukomanovic D, Almulhim M. Emergency department approach to QTc prolongation. The American journal of emergency medicine. 2017 Dec 1;35(12):1928-33.
Johnson JN, Ackerman MJ. QTc: how long is too long?. British journal of sports medicine. 2009 Sep 1;43(9):657-62.
Zhang Y, Post WS, Blasco-Colmenares E, Dalal D, Tomaselli GF, Guallar E. Electrocardiographic QT interval and mortality: a meta-analysis. Epidemiology (Cambridge, Mass.). 2011 Sep;22(5):660.
Roden DM. Long-QT syndrome. New England Journal of Medicine. 2008 Jan 10;358(2):169-76.
Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F. Prevalence of the congenital long QT syndrome. Circulation. 2009 Nov 3;120(18):1761.
Moskovitz JB, Hayes BD, Martinez JP, Mattu A, Brady WJ. Electrocardiographic implications of the prolonged QT interval. The American journal of emergency medicine. 2013 May 1;31(5):866-71.
Banai S, Tzivoni D. Drug therapy for torsade de pointes. Journal of cardiovascular electrophysiology. 1993 Apr;4(2):206-10.
Schwartz PJ. Practical issues in the management of the long QT syndrome: focus on diagnosis and therapy. Swiss medical weekly. 2013 Oct 2;143(3940).
Poterucha JT, Bos JM, Cannon BC, Ackerman MJ. Frequency and severity of hypoglycemia in children with beta-blocker–treated long QT syndrome. Heart Rhythm. 2015 Aug 1;12(8):1815-9.
Holland KE, Frieden IJ, Frommelt PC, Mancini AJ, Wyatt D, Drolet BA. Hypoglycemia in Children Taking Propranolol for the Treatment of Infantile Hemangioma. Arch Dermatol. 2010;146(7):775–778.
Hyltén-Cavallius L, Iepsen EW, Wewer Albrechtsen NJ, Svendstrup M, Lubberding AF, Hartmann B, Jespersen T, Linneberg A, Christiansen M, Vestergaard H, Pedersen O. Patients with long-QT syndrome caused by impaired hERG-encoded Kv11. 1 potassium channel have exaggerated endocrine pancreatic and incretin function associated with reactive hypoglycemia. Circulation. 2017 May 2;135(18):1705-19.
Marzuillo P, Benettoni A, Germani C, Ferrara G, D’agata B, Barbi E. Acquired long QT syndrome: a focus for the general pediatrician. Pediatric emergency care. 2014 Apr 1;30(4):257-61.
Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, Cappelletti D. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348(19):1866




